In providing comprehensive care for patients with SCD, it is vital to
have a well-staffed emergency or other acute care facility where patients
can receive initial care during acute complications. The Emergency
Departments (ED) or acute care center and its staff are important components
of the team that provides care for patients with SCD. Because of
the episodic nature of the complications of SCD, and their potential for
rapid deterioration and serious outcome, patients with SCD frequently consult
the acute care facilities. In many instances the ED is the place
where the initial diagnosis of SCD is made, usually after a complication.
Often, the ED is where patients and their famlies are first introduced
to the medical care of a disease about which they may have little or no
knowledge. The reception and treatment given to these patients and
their famlies in the ED to a large extent determine the degree of confidence
that they develop in that hospital and its staff.
An ED in a community with a large population of patients with SCD must
be prepared to handle the usual and recognize the unusual complications
of the disease. While many of the acute complications of SCD have
well defined signs and symptoms, many of them also mimic other conditions.
Conversely, patients with SCD present with the signs and symptoms of other
conditions which may mimic typical complications of SCD. It is important
to consider the possibility of the existence of other conditions not related
to SCD when patients with SCD present with the signs and symptoms of acute
complications. Patients with SCD vary widely in the manifestations
of their disease. It is important to recognize such individual variation
by relying on past medical records, hisory given by family and the patient,
and advice from those familiar with the disease. Like all patients
with chronic disease, patients with SCD may know more about how the disease
affects them and how they respond to therapy than the treating doctors
and nurses.
In a patient suspected of a serious sickling disorder, diagnostic work
should begin in the acute care facility. Pertinent tests include:
1.
Overwhelming Infections in Sickle Cell Disease
a. Bacterial Infection
-
This was the leading cause of death in young children with SCD in the US,
account for 38% of all deaths in patients under 20 years of age prior to
the establishment of penicillin prophylaxis. Strep. pneumoniae sepsis
and meningitis are the most common causes (86%) and H. influenzae is the
second major cause (11%) of infection deaths in that age group.
The combination of penicillin prophylaxis and multi-valent pneumococcal
vaccination (especially a conjugated vaccine) is expected to decrease the
incidence and mortality of pneumococcal bacteremia in children with SCD.
-
SCD-SC is generally felt to be a milder disease than SCD-SS. However,
children under 3 years of age with SC have a risk of bacteremia at a rate
almost half of those with SCD-SS (Ref #2, below). In general, children
with SCD-SC should be managed with the same degree of caution with regard
to infection as those with SCD-SS.
-
Children with SCD-S
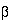 0
are
considered to be of the same clinical severity as those with SCD-SS.
However, those with S+
have
the mildest course of the common SCD types. Their infection risk
is probably much less than those with SCD-SS. However, they end up
being managed like the others because of the lack of data on their degree
of risk.
0
are
considered to be of the same clinical severity as those with SCD-SS.
However, those with S+
have
the mildest course of the common SCD types. Their infection risk
is probably much less than those with SCD-SS. However, they end up
being managed like the others because of the lack of data on their degree
of risk.
-
In the first penicillin prophylaxis study (see reference #4, below),
9 or 16 episodes of pneumococcal sepsis were associated with focal infections:
pneumonia (5), meningitis (2), and otitis media (2). It is incorrect
to assume that the child with a focus of infection is in less danger of
overwhelming sepsis.
-
The most important thing to remember about pneumococcal sepsis in young
children with SCD is its rapid, overwhelming course. The 3 deaths
in the penicillin study all occurred within 9 hours of onset of symptoms.
-
Disseminated intravascular coagulation is a common feature of fatal overwhelming
infection in SCD.
-
Deaths have resulted in part from delay in the institution of appropriate
are caused by lack of prior knowledge of the diagnosis of SCD, failure
of parents to recognize or report signs of infection, and failure of medical
personnel to apprecite the potential danger. However, in many instances,
the patient simply succumbs due to the overwhelming course of the infection,
in spite of appropriate care.
Fever and Infection
Fever is the only reliable indicator of potential infection
in patients with SCD. There are no quick laboratory tests available
which can rule out all bacterial infection or pneumococcal infection alone.
All febrile children with SCD may have serious bacterial infection.
Work-up and management should depend on clinical evaluation considering
age, specific hemoglobinopathy, and physical examination.
Laboratory studies
-
CBC with differential, reticulocyte count, and platelet count.
-
Cultures: Blood and urine; CSF and throat if indicated by clinical
evaluation.
-
Xrays: Chest x-ray, as indicated.
Acute chest syndrome chould be ruled out in any young child who presents
with fever. Pulmonary symptoms may be minimal or absent.
-
CSF culture: Lumbar puncture should be performed based on clinical
judgement. The younger the child with high fever, the stronger the
indication. In a patient with vertebral pain, a stiff neck may mimic
signs of meningitis. If such a patient also has high fever, it is
safer to rule out meningitis with a lumbar puncture.
Management
-
Recent developments have resulted in changes in the standards of practice
in the management of the child with SCD and fever.
-
Traditionally, all such children were admitted for intravenous antibiotics
unti bacterial septicemia was ruled out. The use of the long acting
broad spectrum antibiotic, ceftriaxone, for outpatient management of SCD
children with fever and who do not look ill is gaining popularity.
-
Intravenous broad spectrum antibiotic therapy should be started without
delay (in the acute care facility) in young children, especially those
under five years of age, suspected of serious infection, after appropriate
specimens have been obtained for culture.
-
Admit all febrile children with SCD who look ill for intravenous antibiotic
therapy until bacterial infection has been ruled out. Remeber that
penicillin-resistant pneumococcus is becoming an increasingly common organism.
Children who look ill should be covered for such organisms until cultures
and antibiotic sensitivities prove such coverage unnecessary.
b. Malaria
Malaria is the leading cause of death in children with SCD
in parts of the world where malaria is endemic. In those parts of
the world, prompt diagnosis oand treatment of malaria is a critical part
of the management of the febrile child with SCD. Providers of care
for recent immigrants from malarial regions should be aware of the potential
danger of malaria in these patients.
References
-
Powers, D.R. Natural History of sickle cell disease -- the first
ten years. Semin Hematol 12:267 (1975).
-
Zarkowsky, H., et al. Bacteremia and sickle hemoglobinopathies.
J Pediatr 109:570-585.
-
Gaston, M.H., et al. Prophylaxis with oral penicillin in children
with sickle cell anemia. A randomized trial. N Engl J Med
314:1493 (1986).
-
Wilimas, J., et al. A randomized study of outpatient treatment with
ceftriaxone for selected febrile children with sickle cell disease.
N Engl J Med 329:472-6 (1993).
-
Rogers, Z.R., Morrison, et. al. Outpatient management of febrile
illness in infants and young children with sickle cell disease.
J Pediatr 117:736-739 (1990).
-
Norris, C.F., Mahannah, S.R., Smith-Whitley, K, et al. Pneumococcal
colonization in children with sickle cell disease. J Pediatr 129(6):821-827
(1996).
2. Acute Chest Syndrome
(ACS)
This term has been used to define a relatively common complication of
SCD which may be caused by pulmonary infarction, pneumonia or both.
ACS is defined simply as a new pulmonary infiltrate on chest x-ray or lung
scan. It is usually accompanied by chest pain, respiratory distress,
fever and pleural effusion. It is not uncommon for ACS to develop
or become apparent in patients who have been admitted for management of
other complicationnnnns of SCD, especially painful episodes.
Presentation
-
While ACS seldom presents as an emergency, it always has the potential
for rapid progression with potentially fatal outcome.
-
ACS is often discovered as part of routine evaluation of fever in a child
with SCD.
-
Patients with extensive pulmonary involvement often present with fever,
chest and pleuritic pain, nasal flaring, and abnormal pulmonary auscultation.
There may be rales, rhonchi, and wheezing; often the only abnormality is
decreased breath sounds over a particular area.
-
Rib infarction may be associated with or may precipitate ACS.
-
Patients with pain elsewhere but who show signs of even mild chest discomfort
should be evaluated for possible ACS.
Laboratory Studies
-
Chest x-ray
-
CBC, differential, reticulocyte count
-
Periodic measurement of O2 saturation at steady state in patients with
SCD is a good idea since it may be a useful tool for the diagnosis and
monitoring of the course of ACS. In a study at CHOP of the Hb-O2
saturation in children with SCD, acutely ill patients with transcutaneous
O2 saturation < 96% and a > 3 percentage point decrease from steady
state values, had a probability of having or developing ACS [Reference
#3, below].
-
Patients with moderate respiratory distress deserve arterial blood gas
measurements to help determine their need for oxygen and/or transfusion
therapy.
-
Cultures, as for investigation for bacterial infection. Assessment
for mycoplasmal or chlamydial infection is not done routinely as part of
the initial screening but may be useful if easily available.
Management
-
ACS is treated first as bacterial pneumonia with broad spectrum antibiotics
and oral erythromycin because bacterial infections could potentially be
rapidly progressive in SCD patients.
-
ACS is also managed as a vaso-occlusive/infarctive process with hydration
and analgesics. The possible role of increased intravenous hydration
and narcotic analgesia as contributing factors in the rapid progression
of pulmonary infarction often seen in ACS has been suggested; hydration
should be kept at reasonable but not necessarily high rates. A careful
balance should be maintained between using analgesics to prevent splinting
and permit adequate ventilatory effort, and inducing respiratory depression.
-
Even though thrombosis of pulmonary vessels may be involved in ACS, the
condition has not been treated wtih thrombolytic or anticoagulant therapy.
-
Respiratory distress and documented hypoxia are managed with oxygen therapy
and transfusion of relatively fresh, packed, red blood cells (< 5 day
old bank blood).
-
It is the impression of many clinicians that patients with pulmonary infarction
as the primary basis for ACS tend to respond more readily to red cell transfusions
than those whose ACS may be due primarily to infection.
-
There are preliminary data to suggest a positive role for bronchodilators
in the management of ACS.
References
-
Vichinsky, et al. Causes and outcomes of the acute chest syndrome
in sickle cell disease. NEJM 342:1855-1865 (2000)
-
Castro, O, et al (CSSCD): The acute chest syndrome in sickle cell disease:
incidence and risk factors. Blood 84:643-649 (1994)
-
Rackoff, W.R., Kunkel, N., Asakura, T., Silber, J.H., and Ohene-Frempong,
K. Pulse oximetry and factors associated with hemoglobin desaturation
in children with sickle cell disease. Blood 81:3422-3427
(1993)
-
Vichinsky, E.P., Styles, L.A., Colangelo, L.H., et al. Acute chest
syndrome in sickle cell disease: clinical presentation and course.
Cooperative Study of Sickle Cell Disease. Blood 89(5):1787-1792
(1997)
3. Acute Splenic
Sequestration (ASS)
ASS is a sudden pooling of large amounts of blood into the spleen leading
to acute splenomegaly, profound anemia, and in severe cases, hypovolemic
shock. Death may occur in a few hours.
-
ASS also occurs prior to "autoinfarction" of the spleen, that is, in young
children (usually less than 3 years old) with severe forms of SCD, but
in older children and adults with milder forms such as SCD-SC. Patients
on transfusion or other therapy which may rejuvenate the spleen or delay
its infarction can also develop ASS at older age.
-
ASS often occurs in association with a viral or bacterial infection.
ASS is also seen in association with malaria in children who may or may
not have SCD.
-
Typically, ASS resolves within hours or a few days after a blood transfusion.
-
ASS may be recurrent ("yo-yo" spleen) or may be slowly progressive in development.
-
Splenic sequestration is not always acute. Splenomegaly associated
with more severe anemia than expected for the patient may be evidence of
chronic splenic sequestration which presents with other features of chronic
hypersplenism. Such patients may also have recurrent acute exacerbations.
Presentation
-
Patients may present with signs of acute circulatory insufficiency such
as pallor (check conjunctiva in dark-skinned patients), lethargy, tachypnea,
or shock with massive, tender spleens.
-
It helps to find out the size of the spleen at the last examination for
comparison. Parents who have been taught to measure the size of the
spleen can be helpful to the examiner in determining changes.
Laboratory Studies
-
CBC with differential, reticulocyte count, platelet count
-
Findings include severe anemia with increased reticulocytes and nucleated
RBCs (in contrast with erythroblastopenia); increased WBCs (25,000 - 30,000
range) with a left shift; and decreased platelet count due to hypersplenism.
-
Remeber to interpret the results of the CBC correctly in patients with
milder forms of SCD such as SCD-SC, whose baseline Hbg level may be close
to "normal". Hgb of 6.5 in a teenager with SCD-SC and enlarged spleen
may signify severe sequestration.
-
Cultures as indicated.
-
Type and crossmatch for blood.
Management
-
Prompt restoration of blood volume with plasma expanders is the most important
aspect of emergency management.
-
Correction of anemia should be approached carefully to avoid potential
circulatory overload. In ASS, the spleen has a tendency to shrink
and decrease the volume of the sequestered blood in response to blood transfusion,
thereby reducing the degree of anemia. The goal of initial transfusion
should not be to restore the hematocrit (Hct) or Hgb to normal or steady
state values, but to prevent shock and heart failure. Transfusion
should be performed in gradual steps:
-
For Hct < 12% (Hgb of 4 g/dL), give enough red cells to only double
the Hct after the first transfusion.
-
After initial transfusion, wait for 3 to 4 hours for circulatory equilibration
and check the blood count to establish a new baseline before giving another
transfusion.
-
Look for shrinkage of the spleen during or following each transfusion,
a potential source of volume overload.
-
Further management of recurrent ASS may include chronic transfusion therapy
in the very young patient in order to avoid splenectomy before four or
five years of age, and splenectomy for older children and those in situations
where chronic transfusion is impractical.
References
-
Seeler, R.A. and Shwiaki, M.Z. Acute splenic sequestration (ASSC)
in young children with sickle cell anemia. Clinic Pediatr 11:701-704
(1972).
-
Topley, J.M., et al. Acute splenic sequestration and hypersplenism
in the first five years in homozygous sickle cell disease. Arch Dis
Child 56:765 (1981).
-
Pearson, H.A., et al. Transfusional reversible, functinoal asplenia
in young children with sickle cell anemia. N Engl J Med 283:334
(1970).
-
Michel, B., et al. A fatal case of acute splenic sequestration in
a 53-year old woman with sickle-hemoglobin C disease. Am J Med
92:97-100 (1992).
4. Cerebrovascular
Accident (CVA)
Cerebrovascular accidents (strokes) are among the leading cause of death
and morbidity in children with SCD in the U.S. accounting for 12% of deaths
in patients under 20 years of age. Children with SCD have a risk
of stroke approximately 500 times over that of the general childhood population.
In U.S. urban centers with large African-American populations, SCD is the
leading cause of stroke in children. Strokes occur in about 6 % of
all patients with SCD with an annual incidence of about 1 in 200 patient
(0.5%) but is most common in SCD-SS. Strokes are broadly classified
into two groups, hemorrhagic or thrombotic (infarctive). In children
infarctive strokes are more common.
-
Sometimes fatal in children, CVA in patients with SCD requires emergency
management for stabilization of vital signs and prevention of progression
of neurological damage. Patients have made dramatic recovery from
severe presenting symptoms including coma.
-
Most children recover from CVA with no motor deficits. In the CHOP
series, children <5 years of age are more likely to have residual motor
deficits after stroke than older patients. Degree of neuropsychological
damage and its recovery following CVA has not been carefully assessed.
-
Because CVAs have a high tendency to recur, referral for long term management
is essential.
Presentation
Hemiparesis is the most common presenting symptom of CVA. Patients
also present with transient ischemic attacks, cranial nerve palsies, mono-
or quadraparesis, coma, or seizures. Mild transient episodes in young
children are probably missed. A painless limp in a child with SCD
may be a sign of a stroke.
Laboratory Studies
-
CBC with differential, reticulocyte and platelet count. Strokes have
occurred in patients with severe anemia due to erythroblastopenia (aplastic
crisis) and following excessive transfusion.
-
PT, PTT
-
Cultures, as indicated. Lumbar puncture should not be performed unless
brain imaging reassures against the danger of brain herniation.
-
Blood chemistries, electrolytes.
-
Type and crossmatch for blood.
Management
-
Emergency life support if necessary
-
Seizure treatment if necessary
-
Hydration: Intravenous hydration alone may reverse neurological symptoms.
Avoid excessive hydration because of the danger of cerebral edema.
-
Alert neuroradiology. Brain imaging studies, where available, are
performed as soon as possible after the patient is stabilized to rule out
hemorrhage (which may require surgical intervention) and to define the
lesion. Magnetic resonance imaging and angiography (MRI and MRA)
may be superior to computerized tomography (CT) in defining the pathology
in stroke during the acute period.
-
Early simple and/or exchange transfusion may increase oxygen delivery to
the ischemic site, help restore neurological function, and reduce the potential
for further vaso-occlusion.
-
Long term management to rehabilitate the patien and prevent recurrence
of stroke is an essential followup to acute care.
-
Routine transcranial doppler (TCD) screening of SCD-SS children for evidence
of cerebrovascular disease is now part of standard care. Chronic
transfusion therapy is recommended for children with repeatedly abnormal
TCDs.
Reference
-
Powars, D., et al. The natural history of stroke in sickle cell disease.
Am J Med 65:461-471 (1978).
-
Russell, M.O., et al. Effects of transfusion therapy on arteriographic
abnormalities and on recurrence of stroke in sickle cell disease.
Blood 63:162 (1984).
-
Ohene-Frempong, K. Stroke in sickle cell disease: demographic, clinical
and therapeutic considerations. Semin Hematol 28:213-219
(1991).
-
Adams, R.J., McKie, V.C., Hsu, L., et al. Prevention of first stroke
by transfusions in children with sickle cell anemia and abnormal transcranial
Doppler ultrasonography. N Engl J Med 339:5-11 (1998).
5. Acute Red Cell Aplasia
Severe anemia due to transient cessation of erythropoesis occurs in
all chronic hemolytic diseases. Such acute erythroblastopenias often
associated with infection and B19 parvovirus has been found to be the causative
agent in most cases in the U.S. and elsewhere. Erythroblastosis coupled
with the markedly shortened life span of sickle red cells (16-20 days)
leads to very severe and life threatening anemia in only a few days.
Presentation
-
Patients often present with signs of severe anemia -- pallor, irritability,
headache, or lethargy. Severe constant headache is probably the most
common presenting complaint in children old enough to report headache.
-
In many cases the condition is discovered during evaluation of a child
who has a febrile illness or one with no complaint at a routine visit.
Laboratory Studies
-
CBC with differential, reticulocyte count and platelet count. A low
hemoglobin level with low reticulocyte count, usually less than 1%, is
a hallmark of so-called "aplastic crisis". This is in contrast to
acute splenic sequestration in which severe anemia is accompanied by an
outpouring of young red cells and white cells.
-
Cultures, as indicated.
-
Type and cross match for blood.
Management
-
If the anemia is severe, admit the patent for observation to decide when
or whether to transfuse.
-
The decision to transfuse should be based first on clinical presentation.
The degree of anemia, reticulocyte count, and if available, the state of
erythropoesis in the marrow should be considered in the decision on transfusion.
The goal of initial transfusion is to prevent cardiac failure and shock.
-
When transfusing use small aliquots of red cells to prevent circulatory
overload.
-
B19 parvovirus has been reported to cause first and mid-trimester abortions
due to hydrops fetalis. It is important to isolate patients suspected
of aplastic crisis, especially from pregnant women (including caregivers
such as doctors, nurses and medical students).
References
-
MacIver, J.E., and Parker-Williams, E.J. The aplastic crisis in sickle
cell anemia. Lancet 1:1086-1089 (1952).
-
Serjeant, G.R., et al. Outbreak of aplastic crisis in sickle cell
anemia associated with parvovirus-like agent. Lancet 2:595
(1981).
-
Young, N. Hematologic and hematopoetic consequences of B19 parvovirus
infection. Semin Hematol 25:159 (1988)
C. Painful Episodes
Painful episodes are the most common complication of SCD. This well-known
fact not withstanding, patients with SCD presenting with acute pain often
meet resistance in receiving adequate attention to their pain. Part
of the reason for such reception is the difficulty in assessing a complication
which has no reliable laboratory measurement and often, no objective physical
findings. Painful episodes remain one of the least well-managed complications
of SCD. It is worth remembering that patients do not consult the
acute care facility or their doctors in 70% of painful episodes.
In the Cooperative Study of SCD (Ref #1, below), 39% of 3576 SCD patients
observed for 18,356 patient-years had no episode of severe pain requiring
ED visit or hospitalization; another 40% had less than one painful episode
per year, and 1% had more than six episodes per year. The 5.2% of
patients who had 3-10 episodes per year accounted for more than 32.9% of
all painful episodes.
The pain is usually in the extremities, ribs, sternum, vertebral column
or abdomen.
Patients with recurrent attacks of pain are difficult to manage in the
ED; a well-defined plan of management should be developed to avoid inconsistency.
Presentation
-
Patients complain of "typical crisis pain". Older patients can usually
distinguish between "crisis pain" and other sources of pain.
-
Swelling, erythema, and warmth may or may not be present at the site of
the pain. Do not assume that all pain in a patient with SCD is caused
by acute vasoocclusion. For example, abdominal pain due to cholecystitis
has a different set of evaluation and management options. Also, pain
may be due to causes other than those typical of SCD. It is important
to physically establish the source of pain when examining a young child;
a limp may be painless (see CVA above) and not necessary a sign of a painful
leg.
-
Mild to moderate fever is not an uncommon feature of painful episodes.
Laboratory Studies
-
CBC with differential, reticulocyte count, platelet count. Blood
counts are usually unchanged from steady state levels in uncomplicated
vasooclusive events.
-
Cultures, as indicated.
-
Chest X-ray as indicated. Acute chest syndrome is often associated
with painful episodes; it should be ruled out in patients with chest pain
or fever and pain elsewhere.
Management
The primary goal of management of painful episodes is to alleviate pain
and suffering. Since painful episodes are usually not life-threatening
in children, it is important also to reduce the level of anxiety in the
patient and family. The word "crisis" tends to add to the heightened
anxiety.
Analgesics
Assess the degree of suffering, and select an appropriate drug and dose
based on age and prior history. Acetaminophen, opioids, and nonsteroidal
antiinflamatory drugs (NSAIDS) are the standard medications used in pharmacological
management of sickle cell related pain.
Placebos have no place in the management of sickle cell related pain.
Severe to Moderate Pain
-
Give opioid analgesic, e.g., Morphine sulfate, 0.15 mg/kg/dose q 3-4 hours
IV by infusion over 10-15 minutes or, by subcutaneous injection or intramuscular
injection. IM injections hurt, particularly in children, and lead
to muscle sclerosis.
Where available, patient controlled analgesia (PCA) may be a useful
and less contentious way of administering opioids to patients who are familiar
with its use from prior inpatient management.
Alternative: Ketorolac (Toradol), 1 mg/kg load, then 0.5 mg/kg
q 6 hours.
- Also give NSAIDs, e.g., Ibuprofen 10 mg/kg/dose q 6-8 hours PO. Alternative:
Acetaminophen 10 mg/kg/dose po q 4 hours.
-
If the pain is relieved for 3-4 hours, give effective PO opioid, e.g:
Hydromorphone: 0.05-0.06 mg/kg/dose q 4 hours or
Oxycodone 5-10 mg/dose q 4 hours (adult) or
Codeine 1 mg/kg/dose q 4 hours
and observe for one hour.
Continue NSAID or acetaminophen.
-
If moderate or severe pain returns, repeat the parenteral narcotic dose
and observe.
-
When pain is under control, resume management with oral opioids, as above.
-
If relief is maintained, discharge the patient with a small prescription
and referral for follow-up.
-
A final dose of parenteral narcotics "for the road" is not recommended.
-
If significant pain persists after several hours, admit the patient to
the hospital.
Mild Pain
-
Give NSAID plus acetaminophen alone or in combination with PO opioid as
above. Aspirin can be given if the child has no symptom of upper
respiratory infection.
Note: prevention of drug addiction should not be the reason to withhold
treatment for pain. Patients have become addicted from medications
obtained from outside regular medical channels or from physicians who were
too liberal with prescriptions for home use.
Hydration
-
Clinical experience suggests that increased hydration may shorten the duration
of painful episodes and may make the patent feel more comfortable.
-
Increased oral fluids are adequate for children with mild pain.
-
Intravenous hydration is appropriate for patients with more severe pain,
especially older children and those unable to take increased oral fluids.
-
Administer IV (+PO) fluids at 1.5 x maintenance plus losses, if any.
D5 0.22 NaCl (quarter NS) for young children
D5 0.45 NaCl (half HS) for adolescents and adults
Oxygen
-
Oxygen therapy serves no useful purpose in non-hypoxic patients; prolonged
use may suppress red cell production in the marrow.
-
It does not shorten the duration, reverse or decrease the severity of painful
episodes